






DNA Alignment Software | Features
DNA Alignment functions
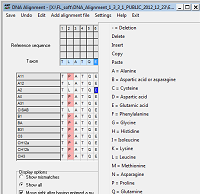
- Aligns both DNA or amino acid data.
- Reads formats: FASTA (*.fas, *.txt), TemporaryAlignment (*.ali).
- Aligns 1 or multiple sequences under a reference sequence.
- Displays alignments and allows editing. With highlighting and motif finding functionality.
- Optional FASTA-import without auto-alignment (i.e. for already aligned data).
- Optional merging of alignment file (*.ali) into current session.
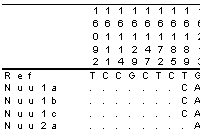
- Writes formats: Network (*.rdf, *.ami), publication-style tables (*.txt), Sequential Nexus (*.nex), Sequential Phylip (*.phy), FASTA (*.fas, *.txt), TemporaryAlignment (*.ali).
Specifications
- Nucleotide position numbering of the reference sequence, including multiple np-ranges in the numbering, is configurable. Example numbering: "15996-16569; 1-576"
- Maximal sequence length 99999.
- Maximal number of sequences unlimited except by available memory on your computer. NB: Under Windows there is a virtual memory limit which users with huge data sizes could hit, but in practice run time duration will be the first limit.
- Hardware requirements: PCs with Windows 10/8/7/XP. Alternatively e.g. Mac with Parallels Desktop and Windows or Oracle VirtualBox/Windows. Alternatively Linux with WINE windows emulation or VirtualBox/Windows. Recommendations: RAM 512MB, CPU 1.5GHz or faster. DNA Alignment can run from memory stick - no Windows registry entries and no Windows Administrator required for installation.
- The alignment algorithm is designed for closely related sequences without large-scale re-arrangements or duplications. Pairwise alignment of each sequence with respect to a user-defined ("reference") sequence. The algorithm is a very sophisticated pairwise alignment algorithm based on the idea in [Morgenstern B et al. DIALIGN: finding local similarities by multiple sequence alignment. Bioinformatics. 1998;14(3):290-4] with specific improvements in our implementation. Unlike older methods, large segments of sequences are compared (a parameter allows user control over the smallest segment size), and there is no "gap penalty". If more than 1 sequence is aligned, the pairwise alignments with the reference sequence are optimised by comparing multiple sequences.
Latest version of DNA Alignment: Version 1.3.3.2 for Windows 10 released on 18 January 2020
DNA Alignment © Copyright Fluxus Technology Ltd 2003-2020